
au sommaire


Cellules souches embryonnaires de souris. Crédit PD-USGov-NSF.
La résolutionrésolution de la structure de l'ADN puis l'initiation de l'étude des mécanismes permettant aux gènes de s'exprimer ont été des avancées majeures de la biologie. Dans le même temps, on découvrait de plus en plus de gènes mais leur rôle demeurait difficile à étudier. Au début des années 1980, on savait replacer les produits d'un gène dans des voies de signalisation que l'on ébauchait avec les techniques de biologie moléculaire; on savait étudier le patron d'expression des gènes dans un organisme grâce à la technique d'hybridationhybridation in situ des ARN et c'était à peu près tout.
Un moyen d'étude qui faisait particulièrement défaut était la possibilité de modifier sélectivement un gène donné afin d'analyser les phénotypes anormaux induits par la mutation et par là, d'en déduire la fonction des gènes. C'est dans la mise en place de cette mutagénèse dirigée (chez la souris) que prennent place Evans, Smithies et Cappecchi qui ont reçu le prix Nobel de médecine cette année.
En 1981, Evans (et Kaufman) parviennent à isoler une lignée de cellules souchescellules souches à partir d'embryonsembryons pré-implantatoires. Cette avancée était intéressante en soi mais c'est Bradley, en 1984, qui va lui donner toute sa portée en parvenant à démontrer la capacité de ces cellules à coloniser un blastocyte et à participer ainsi à la formation d'une souris chimèrechimère. L'intérêt n'apparaît pas immédiatement mais ce qu'il faut bien intégrer c'est que l'on dispose alors d'un outil permettant d'introduire le patrimoine génétiquegénétique d'une cellule issue d'une souris dans une autre souris. L'année suivante (1985), Smithies parvenait à réaliser la première mutagénèse dirigée par recombinaisonrecombinaison homologue atteignant spécifiquement le gène de la bêtabêta-globine dans une cellule de mammifèremammifère. La recombinaison homologue était un événement déjà connu chez les procaryotesprocaryotes mais aussi chez les eucaryoteseucaryotes où elle est à l'œuvre durant les phénomènes de crossing-overcrossing-over durant la méioseméiose.
La nouveauté aura été de réussir à introduire le gène muté par génie génétiquegénie génétique dans la cellule et à sélectionner les cellules ayant vécu cet événement extrêmement rare de recombinaison homologue. Un peu plus tard, en 1987, Capecchi parvenait à muter le locuslocus Hprt dans les fameuses cellules souches de Evans par la technique de mutagénèse par recombinaison homologue développée par Smithies. Il ne restait plus qu'à transférer ces cellules dans un blastocyte. C'est ce que réalisa Koller en 1989 produisant finalement la première souris mutée par recombinaison homologue (pour la petite histoire elle constitue un modèle pour la maladie de Lech-Nyhan). Donc, au début des années 1990, on disposait de cet outil permettant d'étudier la fonction des gènes.
On mute ce que l’on veut !
Le principe de recombinaison homologue était déjà bien connu avant que Capecchi ne l'exploite dans les cellules souches de souris. Il repose sur le fait que quand deux fragments d'ADN sont très semblables, homologues sur au moins une vingtaine de bases, et si une cassure survient, la cellule la répare mais peut se tromper » et ponter les deux morceaux, les recombiner. C'est ce que l'on appelle la recombinaison homologue. L'événement est rare.
Mais un événement encore plus rare peut survenir, c'est la double recombinaison homologue. C'est cet événement qui nous intéresse pourtant ici car c'est lui qui permet d'introduire une nouveauté dans un gène. On comprend bien qu'à partir du moment où l'on dispose d'une séquence qui possède deux régions d'homologiehomologie, peu importe ce qui se trouve entre les deux, si une double cassure survient, il peut y avoir une recombinaison. C'est cette propriété qui a été utilisée par les scientifiques pour muter spécifiquement un gène de la manière dont ils le désiraient. On savait que l'on pouvait injecter de façon transitoire un fragment d'ADN dans une cellule. L'idée était d'utiliser comme fragment une version mutée du gène que l'on ciblait, on savait que statistiquement se produirait dans l'une ou l'autre des cellules un événement de double recombinaison qui introduirait la mutation dans le génomegénome de la cellule. Toute la question était de savoir comment retrouver ces cellules.
La constructionconstruction des fragments d'ADN injectés dans la cellule a alors été pensée pour permettre une sélection en deux temps. En plus du gène muté, on ajoute un gène de résistancerésistance à un antibiotiqueantibiotique (appelé cassette NEO, il confère une résistance à la néomycine), ainsi qu'une cassette TK (TK pour thymidinethymidine kinasekinase, confère une sensibilité au gancyclovir). Le gène de résistance sert à trouver les cellules qui ont bien reçu le transgènetransgène : les cellules sans transgène meurent en présence de néomycine (figure 1). Ainsi on pourra les laisser se multiplier et augmenter les chances de survenue de la double recombinaison. La cassette TK permet de tester si la recombinaison a eu lieu. On sait déjà que les cellules ont intégré le transgène. La cassette TK n'étant homologue à aucun gène de la cellule, elle n'est jamais intégrée au génome. Une fois que la recombinaison a eu lieu, en principe, le transgène est dégradé. Les cellules perdent donc le gène de sensibilité. Dès lors, seules les cellules n'ayant pas recombiné sont sensibles au gancyclovir. On retrouve ainsi les cellules ayant intégré la mutation dans leur génome.

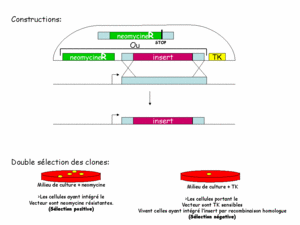
Cliquez pour agrandir
Figure 1: constructions et sélections des clones recombinants.
Il ne reste plus qu'à intégrer ces cellules dans un blastocyte et attendre que les chimères naissent. On appelle chimère, les souris dont les organes sont formés de ses cellules et des cellules issues de la cellule souche que l'on a injectée. En croisant ces souris avec une autre souris sauvage on peut voir naître des souris hétérozygoteshétérozygotes pour le gène muté. En croisant cette première génération on peut obtenir des souris homozygoteshomozygotes.
Voilà pour le principe de base.
Cette technique de mutagénèse dirigée chez la souris a suscité immédiatement de grands espoirs. On pouvait inactiver spécifiquement un gène pour en étudier la fonction. Si les premiers essais se sont plutôt portés vers les domaines de la cancerogenèse comme par exemple l'inactivation du fameux gène P53P53, les plus grands pourvoyeurs de souris mutantes, ou encore, knock-down (KO) furent rapidement les embryologistes. La possibilité d'étudier les phénotypes des souris mutantes a permis une avancée considérable de la biologie du développement. La réalisation de mutations ciblées n'a toutefois pas pour unique but d'inactiver une fonction. Au contraire, on souhaite parfois la conserver. Un exemple très intéressant est celui des souris non plus KO mais knock-in. Le gène est toujours inactivé mais il l'est par l'insertion de la séquence codant pour la GFP (green fluorescent proteingreen fluorescent protein)) dans la séquence codant pour la protéineprotéine. Comme on peut l'imaginer ceci a pour effet de suivre le devenir des cellules dans l'organisme. Si le gène n'est pas dose dépendant, c'est-à-dire que sa fonction peut être assurée avec une seule copie, alors les hétérozygotes permettent de savoir quelles cellules expriment le gène et à quel moment.
Reste que l'utilisation de souris mutantes a donné lieu à certaines désillusions qu'il a fallu expliquer. Par exemple, la carencecarence fœtale en vitamine Avitamine A conduit à un syndromesyndrome polymalformatif décrit avec précision. Chez la souris, il existe trois récepteurs de l'acideacide rétinoïque (dérivé métabolique actif de la vitamine A) dont la distribution dans les tissus au cours du développement embryonnaire était connue. Dès lors, en fonction des récepteurs mutés, on pensait pouvoir reproduire, au moins partiellement, le syndrome de carence fœtal en vitamine A.
Les résultats furent décevants, les souris ne présentaient presque aucun symptômesymptôme. Ce genre de déconvenues mit en lumièrelumière l'extrême plasticitéplasticité des systèmes au cours du développement. En effet, si un des récepteurs fait défaut, un autre peut prendre sa place et en compenser l'absence. La présence des trois récepteurs est décrite comme une redondance physiologique, phénomène retrouvé depuis dans plusieurs voies de signalisations. Pour voir apparaître un syndrome de carence fœtal en vitamine A, il fallait muter dans la même souris deux des trois gènes codant pour les récepteurs de l'acide rétinoïque.
Parfois, les déceptions furent non pas une absence de symptôme mais au contraire une gravitégravité insoupçonnée. Par exemple, FGF8 est une moléculemolécule de signalisation très importante pour la mise en place des os, des muscles. La mutation du gène codant pour FGF8 n'a jamais permis d'étudier son rôle dans ces événements. En effet, il se trouve que les embryons meurent d'un défaut de gastrulationgastrulation au stade blastocyte, bien avant la mise en place du squelette et des muscles. Une limite apparaissait donc clairement, on ne maîtrisait pas la survenue de la mutation dans le temps. On ne la maîtrisait pas plus dans l'espace. Toutes les cellules de la souris portent la mutation depuis la fécondationfécondation. Il fallait donc trouver une amélioration afin de contrôler la survenue de la mutation. L'utilisation de système cre/loxP du bactériophagebactériophage P1 apportera une première innovation.
On mute où on veut !
Ce système emprunté au bactériophage P1 a permis une réelle avancée dans le domaine de la mutagénèse dirigée. Ce système loxP est un site permettant la recombinaison de l'ADN par une enzymeenzyme recombinase Cre. On ne trouve pas ces sites dans les génomes eucaryotes, il était donc possible de les y ajouter pour apporter une action nouvelle et spécifique que nous allons décrire.
Les sites loxP sont des sites asymétriquesasymétriques de 34 paires de bases, aussi peut-on les orienter sur l'ADN. De plus, s'ils sont insérés en phase dans une séquence codante, alors le peu d'acides aminésacides aminés ajoutés ne perturbent pas la fonction de la protéine ciblée. On peut donc dire que, sous réserve de certaines précautions, l'insertion de site loxP dans une séquence codante n'a aucune incidenceincidence.
Pour pouvoir recombiner l'ADN, la recombinase Cre a besoin de deux sites loxP. Son action dépend alors de l'orientation de ces sites sur l'ADN comme l'illustre la figure 2.

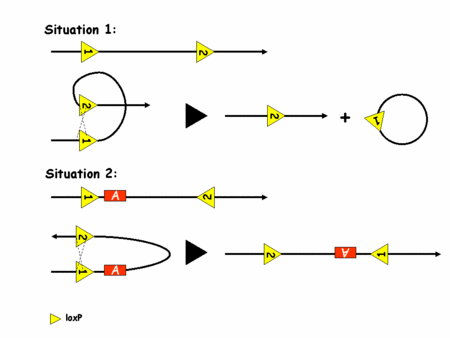
Figure 2: recombinaison de l'ADN par le système Cre/loxP
C'est l'utilisation de deux sites loxP dans le même sens qui a été le plus vite exploité. En effet, on voit bien que dans ce cas là, la Cre excise le fragment d'ADN situé entre les deux sites loxP. On peut donc contrôler la survenue de la mutation en contrôlant l'expression de la Cre. Ceci est rendu possible par de la transgénèsetransgénèse classique (insertion aléatoire d'un fragment d'ADN exogèneexogène dans un génome). Le transgène utilisé est alors la séquence codante de la Cre sous le contrôle du promoteur d'une protéine spécifiquement exprimée dans le tissu qui nous intéresse. Par exemple et grossièrement, on utilisera un promoteur de kératinekératine pour inactiver un gène dans la peau. Pour parvenir à ses fins il suffit alors de générer des souris dont les deux allèlesallèles du gène d'intérêt portent deux sites loxP dans le même sens et des souris porteuses du transgène Cre. En gérant bien ses croisements on finit par obtenir une souris avec les sites loxP et la Cre, ce qui induit la mutation dans le tissu désiré. On a donc un contrôle spatial de la survenue de la mutation. Le contrôle temporel n'est en revanche toujours pas parfaitement maîtrisé. Une nouvelle amélioration va permettre d'y parvenir. C'est le système CreERT (eostrogen receptor tamoxifen).
On mute quand on veut !
L'idée est ici d'utiliser non plus la Cre mais une protéine chimère formée de la Cre et d'un domaine de liaison des ligandsligands (DLL) modifié du récepteur des œstrogènesœstrogènes (ER). Physiologiquement, ER est un récepteur nucléaire présent dans le cytoplasmecytoplasme des cellules qui l'expriment. En présence d'œstrogène, il migre dans le noyau (d'où son nom de récepteur nucléaire) et va se fixer sur l'ADN (grâce à son domaine de liaison de l'ADN) où il activera ses gènes cibles. La translocationtranslocation cytoplasme-noyau est possible grâce au DLL (en haut de la figure 3, ci-dessous).

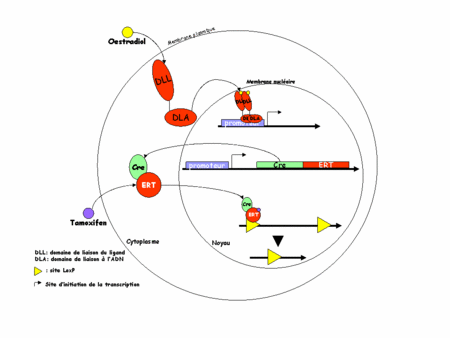
Figure 3 : recombinaison par le système CreERT
Dans la construction CreERT, la chimère n'est construite qu'avec le DLL ceci permet une translocation vers le noyau mais rend impossible toute fixation sur l'ADN, rendant le récepteur inactif. De plus le DLL a été modifié de telle sorte qu'il ne peut plus lier les œstrogènes endogènesendogènes mais uniquement un analogue structural pharmacologique : le tamoxifen (d'où le TT de ERT). On peut alors comprendre le système. En absence de tamoxifen, la chimère CreERT est exprimée dans les cellules cibles mais l'ER non ligandé reste dans le cytoplasme. La recombinaison de l'ADN par la Cre ne peut pas avoir lieu. C'est l'expérimentateur qui choisit quand aura lieu cette recombinaison en injectant dans sa souris du tamoxifen (figure 3, en bas).
C'est ainsi que l'on peut avoir un contrôle spatial (par le choix du promoteur contrôlant l'expression de la Cre) et temporel (par le choix du moment d'injection du tamoxifen) de la survenue de la mutation dans la souris. L'utilisation de ces modèles animaux dépasse à présent largement le simple cadre de la biologie du développement. En cancérologiecancérologie par exemple où la mutation germinale d'un gène ne permettait pas de reconstituer la physiologie des tumeurstumeurs.
On fait ce que l’on veut !
Comme nous l'avons vu les systèmes s'affinaient mais il restait certaines choses que l'on ne parvenait pas à faire. Par exemple, suivre le devenir des cellules mutées, permettre aux cellules de recouvrir un génotypegénotype sauvage après une mutation. Récemment tout ceci a été rendu possible par le développement du système FLEX.

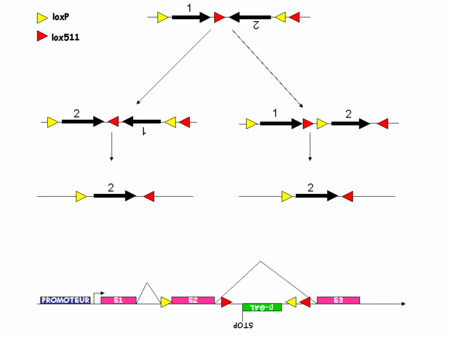
Ce nouveau système exploite le fait que la Cre inverse l'orientation d'un fragment d'ADN flanqué de deux sites lox en position têtes bêches. Il exploite aussi le fait que la Cre ne permet la recombinaison de l'ADN que si les sites lox sont parfaitement identiques. Si deux sites lox présentent des différences alors la Cre ne peut pas agir. Par exemple, la Cre ne peut pas recombiner un fragment d'ADN flanqué d'un coté d'un site loxP et de l'autre d'un site lox511. C'est l'utilisation raisonnée de deux paires de sites lox qui permet de créer une sorte d'interrupteur remplaçant un fragment d'ADN, quel qu'il soit à l'endroit et au moment désiré (figure 4).
Nous avons vu que le génie génétique permet une étude toujours plus fine de la régulation de l'expression des gènes. L'évolution de toutes ces techniques est vertigineuse et l'on peine à mesurer ce qu'il sera possible de faire demain. Reste que si tout ceci est réalisable, c'est au travail d'Evans, de Capecchi et de Smithies que nous le devons et c'est en cela que le prix Nobel de physiologie et de médecine qui leur a été attribué cette année est amplement mérité.
Bibliographie
- Feil R et al. (1997) Biochemical and Biophysical Research Communications. 237, 752-757.
- Ledermann B. Embryonic stem cells and gene targeting. (2000) Experimental Physiology. 85(6), 603-613.
- Schnütgen F. et Ghyselinck. N.B. (2007) Adopting the good reFLEXes when generating conditional alterations in the mouse genome. Transgenic Research 16, 405-413.

par Hugues Jacobs
le 29 octobre 2007









. © Nasa/domaine public")












