
au sommaire


La mucoviscidose est une maladie génétique qui touche environ 1 enfant sur 4.500 naissances et qu'on ne parvient pas à guérir. Un traitement efficace serait donc le bienvenu. © Spooky Pooka, Wellcome Images, cc by nc nd 2.0
La mucoviscidose est une maladie génétique qui touche les épithéliums de nombreux organes, en particulier ceux des poumons, du pancréas et de l'intestin. Dans les poumons, elle se manifeste par une hydratationhydratation insuffisante de l'épithélium qui débouche sur une surabondance de mucus dans les bronches. Ce mucusmucus retient les agents pathogènespathogènes, et favorise les infections et les inflammationsinflammations chroniques qui finissent par être mortelles pour les personnes atteintes.
La maladie est causée par des mutations dans le gènegène codant pour une protéineprotéine appelée CFTRCFTR (Cystic Fibrosis Transmembrane conductanceconductance Regulator)). Cette protéine, essentielle pour le passage de l'eau à travers les épithéliums, est un canal ioniquecanal ionique qui permet aux ionsions chlorure de traverser la membrane des cellules. Actuellement, environ 2.000 mutations du gène conduisant à la maladie sont connues. Néanmoins, 70 % des cas de mucoviscidose sont dus à une seule mutation appelée ΔF508. C'est cette mutation que ciblent deux moléculesmolécules qui viennent d'être découvertes par une équipe internationale de scientifiques, dirigés par Aleksander Edelman (Institut fédératif de recherche Necker - Enfants malades).
 vers le milieu extracellulaire. La localisation de la mutation ΔF508 est indiquée en vert. © Toony, Wikipédia, cc by sa 3.0")
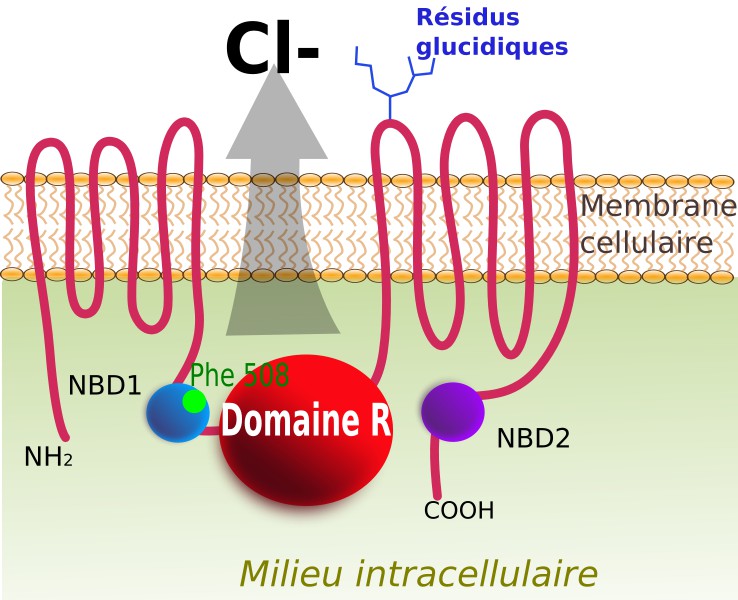
Le récepteur CFTR est schématisé ici. Il joue le rôle d'un canal ionique permettant la sortie des ions chlorure (Cl-) vers le milieu extracellulaire. La localisation de la mutation ΔF508 est indiquée en vert. © Toony, Wikipédia, cc by sa 3.0
La mucoviscidose vaincue in vivo et in vitro
Les chercheurs ont réalisé un criblage informatique sur 200.000 molécules, recherchant celles qui pourraient interagir avec une zone spécifique de la protéine anormale. Ils ont ainsi trouvé une douzaine de molécules potentiellement actives. Avec ces composés, ils ont réalisé des tests in vitroin vitro sur des cultures de cellules humaines, et in vivoin vivo sur des souris présentant cette mutation. Ils ont ainsi observé que deux de ces molécules permettent à la protéine mutée ΔF508-CFTR d'être acheminée à la membrane et de remplir son rôle.
Point fort de ce travail publié dans EMBO Molecular Medicine : les chercheurs ont décrit le mécanisme d'action de ces deux molécules. En soi, la protéine ΔF508-CFTR, malgré sa mutation, pourrait remplir de façon satisfaisante sa fonction. Le problème est qu'une fois synthétisée, elle est reconnue comme anormale par une autre protéine, la kératinekératine 8, qui favorise sa dégradation. Ainsi, ΔF508-CFTR n'atteint pas la membrane cellulairemembrane cellulaire. Les molécules découvertes par les chercheurs font obstacle à l'interaction entre la kératine 8 et ΔF508-CFTR. Ainsi, la protéine peut se déployer convenablement et agir comme canal ionique. Les scientifiques pensent que, dans le cadre d'un éventuel traitement, les deux composés trouvés pourraient être associés à des molécules dites « potentiatrices », qui permettent d'augmenter l'activité de ΔF508-CFTR.
À présent, les chercheurs veulent savoir si, chez les souris modèles de la mucoviscidose, ces deux molécules permettent effectivement de diminuer leur susceptibilité aux infections. Dans les années à venir, ils espèrent aussi commencer les tests cliniques sur des personnes malades.

par CNRS
le 19 septembre 2013


 ; à comparer avec un scanner normal (en bas) où ne sont visibles que quelques ombres vasculaires.")



















