
au sommaire


La drépanocytose, qui vient du mot faucille en grec, porte bien son nom. Les globules rouges ont en effet la forme de cet instrument comme le montre l’image ci-dessus. Cette maladie héréditaire se caractérise par l’altération de l’hémoglobine, la protéine qui transporte le dioxygène dans le sang. Les ADN non codants contrôlent certains symptômes de cette maladie. © Wellcome image, Flickr, cc by nc nd 2.0
Les bêta-thalassémies et la drépanocytose font partie des troubles héréditaires touchant les globules rouges les plus fréquents. La drépanocytose, qui affecte 300.000 nouveau-nés chaque année, est même sur le point de devenir la maladie génétique la plus commune en Europe. Les symptômes sont causés par des mutations dans le gène de la bêta-globine, conduisant à des altérations de l'hémoglobine adulte, responsable du transport de l'oxygène dans le sang. Malgré l'implication d'un gène unique, ces deux maladies peuvent être très sévères. De nombreux facteurs peuvent cependant en modifier la gravitégravité, en particulier la capacité de certains individus à produire de l'hémoglobine fœtale normalement absente chez l'adulte. Chez les patients thalassémiques et drépanocytaires, cette synthèse a un effet bénéfique car elle compense, en partie, les défauts d'hémoglobine adulte.
Les régions non codantes du génomegénome, autrefois appelées ADNADN poubelle, ont un rôle aujourd'hui reconnu dans la régulation des gènes. Les mutations ou variations dans leurs séquences peuvent en effet être impliquées dans la survenue ou la sévérité de certaines pathologiespathologies comme le diabète, les maladies cardiovasculairesmaladies cardiovasculaires ou les cancers. Pourtant, ces variants génétiques, présents en grand nombre dans les régions non codantes du génome, sont en général localisés à des distances considérables des gènes.

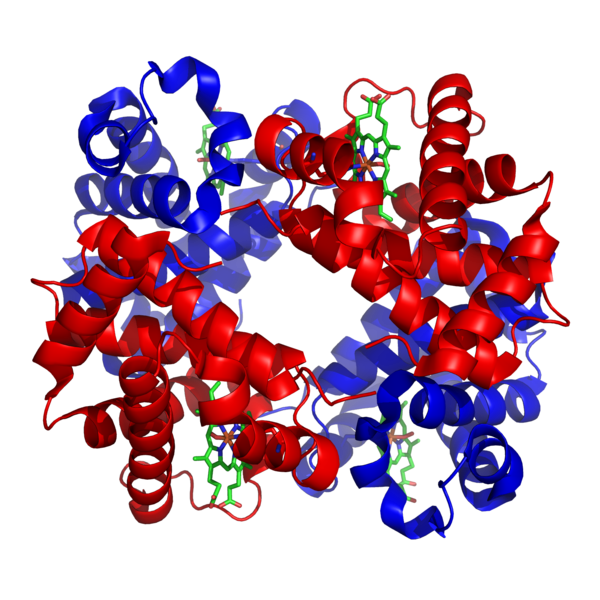
Structure de l’hémoglobine, la protéine responsable du transport de l’oxygène dans le sang. © Zephyris, Wikimedia Commons, cc by sa 3.0
Dans une étude précédente datant de plus de dix ans, une équipe avait identifié des variants génétiques liés à la production d'hémoglobine fœtale chez l'adulte. Leurs mécanismes d'action étaient toutefois restés inexpliqués jusqu'à aujourd'hui. En effet, ces variants ne sont pas localisés sur les chromosomeschromosomes qui contiennent les gènes produisant l'hémoglobine mais se trouvent dans un « désertdésert génétique » du chromosome 6q23, à des dizaines de milliers de paires de bases des gènes les plus proches.
Un contrôle à 80.000 paires de base de distance
Dans une étude récente, publiée dans la revue Journal of Clinical Investigation, des scientifiques de l'Inserm viennent enfin d'éclaircir ce mystère. En combinant l'étude du repliement des chromosomes à des analyses d'ADN à haut débitdébit sur des prélèvements effectués chez des patients thalassémiques, les chercheurs ont élucidé les mécanismes par lesquels les variants non codants améliorent les symptômes des thalassémiesthalassémies et drépanocytoses. Ils ont montré que ces variants interagissaient physiquement avec le gène MYB, distant de plus de 80.000 paires de bases, grâce au repliement des chromosomes. « Il faut imaginer notre ADN comme une pelote de laine. Deux régions très éloignées l'une de l'autre quand elle est déroulée peuvent être très proches au sein de la pelote », explique Eric Soler, le directeur de l'étude.
Cependant, chez les patients atteints de bêta-thalassémiebêta-thalassémie ou de drépanocytose et porteurs de ces variants, les chercheurs ont constaté une diminution des repliements des chromosomes. Les variants accèdent alors plus difficilement au gène MYB et l'activent moins efficacement. Cette baisse d'expression du gène MYB conduit à une réactivationréactivation des globines fœtales saines, normalement silencieuses chez l'adulte, permettant de reconstituer une hémoglobine fonctionnelle. « La diminution de l'expression du gène MYB permet ainsi de compenser le défaut de globines adultes et d'améliorer significativement les symptômes des bêta thalassémies et de la drépanocytose », expliquent les auteurs. « Le gène MYB représente une nouvelle cible thérapeutique majeure pour le traitement des bêta-thalassémies et de la drépanocytose, poursuivent-ils. La réactivation de l'hémoglobine fœtale constitue en effet une stratégie thérapeutique de choix. »




. Cette vision des choses était seulement partielle. © Zephyris, Wikipédia, cc by sa 3.0")


 : quelle est la composition de notre sang ? © Andrea Danti, Shutterstock")


. Ils sont très déformables, ce qui leur permet de passer dans des capillaires sanguins très fins, dont le diamètre est inférieur au leur. © geralt, Pixabay, DP")













