

Plusieurs instituts de recherche publique français ont annoncé l'arrêt temporaire des recherches sur les prions après qu'un ancien agent de l'Inrae a développé les symptômes de la maladie de Creutzfeldt-Jakob.
au sommaire
C'est une décision qui a été prise conjointement par l'Anses, le CEA, le CNRS, l'Inrae et l'Inserm, et appuyée par le ministère de l'Enseignement supérieur, de la recherche et de l'innovation. Les laboratoires français ont suspendu provisoirement - pour trois mois - la recherche sur les prions depuis le 27 juillet 2021.
Une mesure de précaution encouragée par la découverte d'un cas possible de la maladie de Creutzfeldt-Jakob chez un ancien chercheur de l'Inrae qui a travaillé sur les prions. Cette dégénérescence du cerveau peut avoir plusieurs origines : génétique, sporadique ou iatrogène, c'est-à-dire une infection via un instrument contaminé. La forme contractée par l'ancien chercheur n'est pas encore connue.
Ce n'est pas un cas unique. En 2019, une assistante-ingénieur est décédée de la maladie de Creutzfeldt-Jakob. Elle avait été infectée en 2010 à la suite d'une blessure lors d'une expérimentation. L'enquête menée après cet incident a conclu « à la conformité réglementaire des laboratoires visités », écrit l'Inserm dans son communiqué de presse.

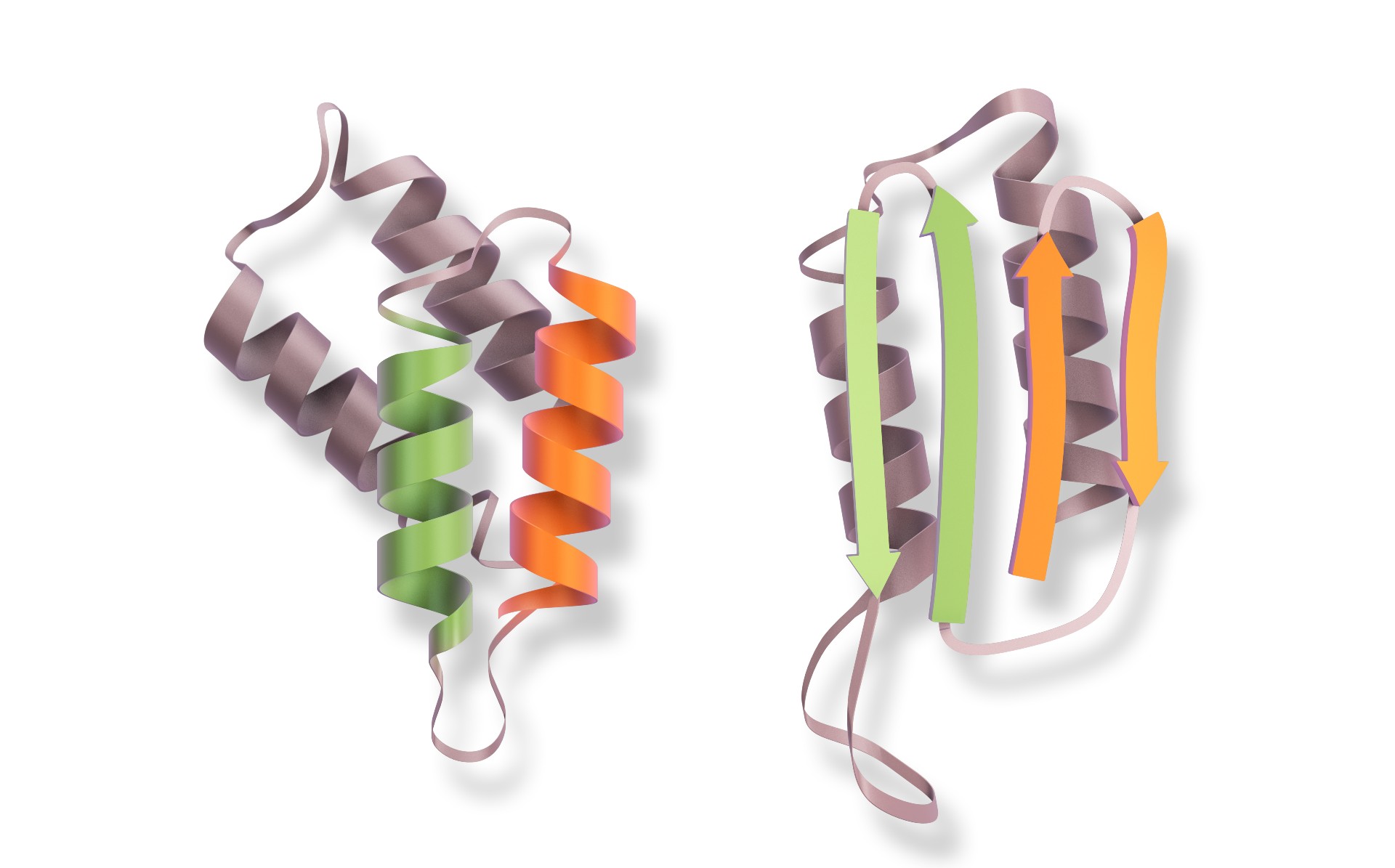
Une protéine comme agent infectieux
L'agent étiologique de la maladie de Creutzfeldt-Jakob est la protéineprotéine prion (PrPc)), une protéine exprimée dans beaucoup de cellules du corps. Dans sa forme saine et active, elle est structurée par des hélices alpha. Mais elle peut aussi prendre une forme pathologiquepathologique, constituée de feuillets bêtabêta. On l'appelle alors PrPcsc ou scrapie.
Sous cette forme, elle s'agglomère dans les cellules et peut transformer les protéines prions saines en scrapie en leur transmettant leur structure anormale. Cet agent infectieux est le seul connu dépourvu d'acides nucléiquesacides nucléiques, les briques constituantes de l'ADNADN et l'ARNARN.
L'accumulation des scrapie est lente et ses effets néfastes apparaissent plusieurs années après la contaminationcontamination. Le diagnosticdiagnostic est difficile et la maladie à prions est souvent confirmée post-mortem. En effet, après la déclaration des premiers symptômessymptômes, la maladie évolue rapidement jusqu'au décès du patient. Aucun traitement spécifique n'existe, les médecins ne peuvent que soulager les symptômes.

 s'accumulent entre les neurones (en bleu) et sont liées à l’agrégation de protéines mal repliées. Le phénomène est tout à fait similaire dans les maladies à prions. © Juan Gaertner, Shutterstock")

, est une pathologie neurodégénérative causée par un prion. Elle est transmissible à l’Homme par la consommation de produits contaminés. Elle a fait 204 victimes humaines, touchées par des symptômes proches de la maladie de Creutzfeldt-Jakob, une maladie de même nature que l'ESB. © matdur69, Flickr, cc by nc nd 2.0")









 ou du câprier ovale (Capparis ovata), avant d'être utilisés comme condiments. Notamment dans la cuisine méditerranéenne. © Fotogiunta, Adobe Stock")










