

au sommaire
Vous êtes biologiste et vous voulez savoir quel est le degré de parenté et l'histoire évolutive entre les différentes espècesespèces de levureslevures (ou de vers, ou d'insectesinsectes, ou d'oiseaux) que vous êtes en train d'étudier. La méthode la plus fiable, celle qui ne vous induira pas en erreur par des ressemblances morphologiques superficielles, c'est la phylogénie moléculairephylogénie moléculaire : comparer le génomegénome de vos bestioles. Pour ce faire, vous disposez désormais de banques de données regroupant des millions de séquences d'ADNADN correspondant à des dizaines de milliers d'espèces vivantes. Mais, sans outils adaptés pour les comparer, elles resteront une suite muette de A, T, C et G.
C'est là qu'interviennent OlivierOlivier Gascuel et Stéphane Guindon, chercheurs au Laboratoire d'informatique, de robotique et de microélectronique de Montpellier (Lirmm). Ces chercheurs ont mis au point en 2003 un puissant algorithme. Il permet d'estimer les relations évolutives liantliant un ensemble d'organismes à partir de la comparaison de leur ADN ou bien des acides aminésacides aminés de leurs protéinesprotéines. Cette méthode a eu un succès fulgurant, tant et si bien que le papier qui la présentait dans Systematic Biology est aujourd'hui en bonne place dans le Science Citation Index.
On comprend pourquoi : cet algorithme permet de réduire drastiquement les temps de calcul par rapport aux méthodes précédentes. De plus, il peut être utilisé avec des données bien plus complexes et longues. « Il y a des recherches que l'on n'imaginait pas pouvoir faire et qui ne demandent maintenant que quelques minutes, explique Olivier Gascuel. Personne n'osait aborder des problèmes qui mettaient en jeu 100, 200 ou 500 espèces et des séquences longues de plusieurs milliers, voire dizaines de milliers de lettres. Après des jours et des jours de calcul on devait tout arrêter sans avoir obtenu de résultat. Notre logiciel permet de traiter ces données. »
Sur la base de cet algorithme, les chercheurs ont développé un logiciel baptisé PhyML. Il est en accès libre sur Internet, comme c'est l'usage dans le domaine. Les chercheurs du monde entier profitent de sa simplicité d'utilisation : ils n'ont plus qu'à entrer leurs données et à attendre que les huit processeurs de l'ordinateur du Lirmm s'en occupent. Ils reçoivent les résultats par e-mail quelques heures ou minutes plus tard. « Le serveur tourne à plein régime : il y a trois ou quatre utilisateurs en permanence. À chaque fois que je vais dans un congrès, j'entends des gens parler de notre programme ! »
Le principe de base de l'algorithme est celui du maximum de vraisemblance, concept majeur en statistique. Il consiste ici à définir un modèle hypothétique qui décrive à la fois le degré de parenté entre les espèces étudiées et les mutations qui ont pu se produire à partir de la séquence originale postulée. Ensuite, il calcule la probabilité que les données correspondent à l'hypothèse de départ. Le processus est réitéré, modèle après modèle, jusqu'à ce que cette probabilité soit maximale. Le modèle correspondant constitue la réponse de l'algorithme. Ce tâtonnement demandait des temps de calculs colossaux jusqu'à l'arrivée de PhyML, qui est en fait une approximation plus que satisfaisante de ce principe. C'est ainsi qu'on voit les espèces former un arbrearbre où chaque noeudnoeud correspond à un ancêtre communancêtre commun et où la longueur des branches représente le temps pendant lequel les espèces ont évolué.

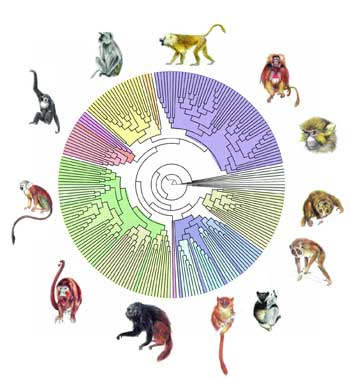
Grâce à PhyML, on a pu reconstruire l'arbre phylogénétique des primates à partir de séquences d'ADN représentant plus de 900 000 paires de bases. Chaque nœud correspond à un ancêtre commun, et la longueur des branches, au temps d'évolution.
© F. Chevenet /IRD.
« Autrefois, on faisait des phylogénies sur un seul gènegène, alors que dans les banques d'ADN on en dispose de milliers. Aujourd'hui, chaque espèce est représentée par 50 ou 100 gènes, et donc, les phylogénies sont beaucoup plus fiables »,explique Olivier Gascuel. Une aubaine pour les biologistes, car la phylogénétiquephylogénétique prend de plus en plus d'importance.
« Environ un papier sur quatre en biologie contient une analyse phylogénétique, rappelle le chercheur. On utilise ces méthodes depuis l'étude fonctionnelle des gènes jusqu'aux recherches sur la biodiversitébiodiversité en passant, par exemple, par l'étude des virus comme le VIHVIH ou le Sras. »
La phylogénétique a provoqué de véritables petites révolutions chez les biologistes systématiciens. Ainsi, on s'est aperçu que dans le monde végétal, les caractères morphologiques comme la forme des feuilles ou la couleurcouleur des fleurs étaient de très mauvais indicateurs pour regrouper les plantes en ordres, familles et genres. En revanche, évaluer leur degré de parenté au niveau du génome est maintenant une méthode très sûre.
Avec PhyML, au grand projet classificatoire initié par Linné, le célèbre naturaliste suédois, s'associent les techniques les plus récentes de l'inférence statistique. Mais un autre intérêt se dégage : la possibilité de comprendre les processus évolutifs au niveau des génomes. Un vaste programme de recherches auquel participent nos deux chercheurs : Olivier Gascuel s'intéresse aux séquences répétées de l'ADN tandis que Stéphane Guindon (entré cette année au CNRS) travaille sur les mécanismes de sélection des mutations chez les virus.
Dans leurs travaux, ils se servent, bien entendu, de leur propre algorithme. Mais aussi des nouvelles versions du programme qu'ils sont en train de développer. Car PhyML n'est pas un fossile vivantfossile vivant destiné à se maintenir tel quel. Les chercheurs travaillent sur des prototypes, des variants qui devraient permettre une sélection plus fiable du bon modèle. Le logiciel évolue... et c'est bien naturel !
Sebastián Escalón
Contact
Olivier Gascuel
Lirmm, Montpellier
[email protected]

par Journal du CNRS
le 3 décembre 2005













. © Dall.E")






