
au sommaire


Le myélome multiple peut s'observer sur une radiographie du squelette : des taches plus sombres indiquent la dégradation de l'os par les tumeurs ostéolytiques. Ici, il s'agit de l'avant-bras d'un patient. © Hellerhoff / Licence Creative Commons
Le myélome multiple (ou maladie de Kahlermaladie de Kahler) est un cancer encore mal connu et toujours incurable. La maladie atteint des cellules de la moelle osseuse, les plasmocytes, qui sont la forme activée des lymphocytes B (producteurs d'anticorps). Elle a pour conséquence la présence d'une grande quantité d'anticorps dans le sang, et est souvent diagnostiquée par une simple analyse sanguine. Des tumeurs ostéolytiques se développent, provoquant la destruction des parties osseuses, fréquemment observables par radiographiesradiographies du squelette, où des zones plus sombres apparaissent.
Les causes de la maladie restent encore floues. Le chlordécone, pesticide toxique, est toutefois montré du doigt dans le nombre significativement plus important des myélomes multiples recensés en Martinique. Les marqueurs génétiquesgénétiques associés à la maladie sont hétérogènes, et jusqu'à présent, les mécanismes moléculaires communs de l'initiation et de la progression du cancercancer chez les différents patients atteints n'étaient pas compris. Un frein dans la découverte d'un traitement efficace et généralisable.
Des chercheurs suédois de l'Université d’Uppsala ont tenté d'identifier le dénominateur commun de la maladie chez différents patients atteints de myélome multiple, et ont publié leurs travaux dans le journal Plos One. L'intégration des génomesgénomes a permis de montrer un lien entre tous ces patients : la sous-expression de plusieurs gènesgènes dans les cellules plasmocytaires, par rapport à des cellules saines de moelle osseuse. Plus la maladie progresse, et plus le nombre de gènes sous-exprimés augmente.
. Le rouge indique une forte expression, le bleu une faible expression. Plus le stade de la maladie est avancé, plus les gènes s'éteignent. © Université d'Uppsala / <em>Plos One</em>")
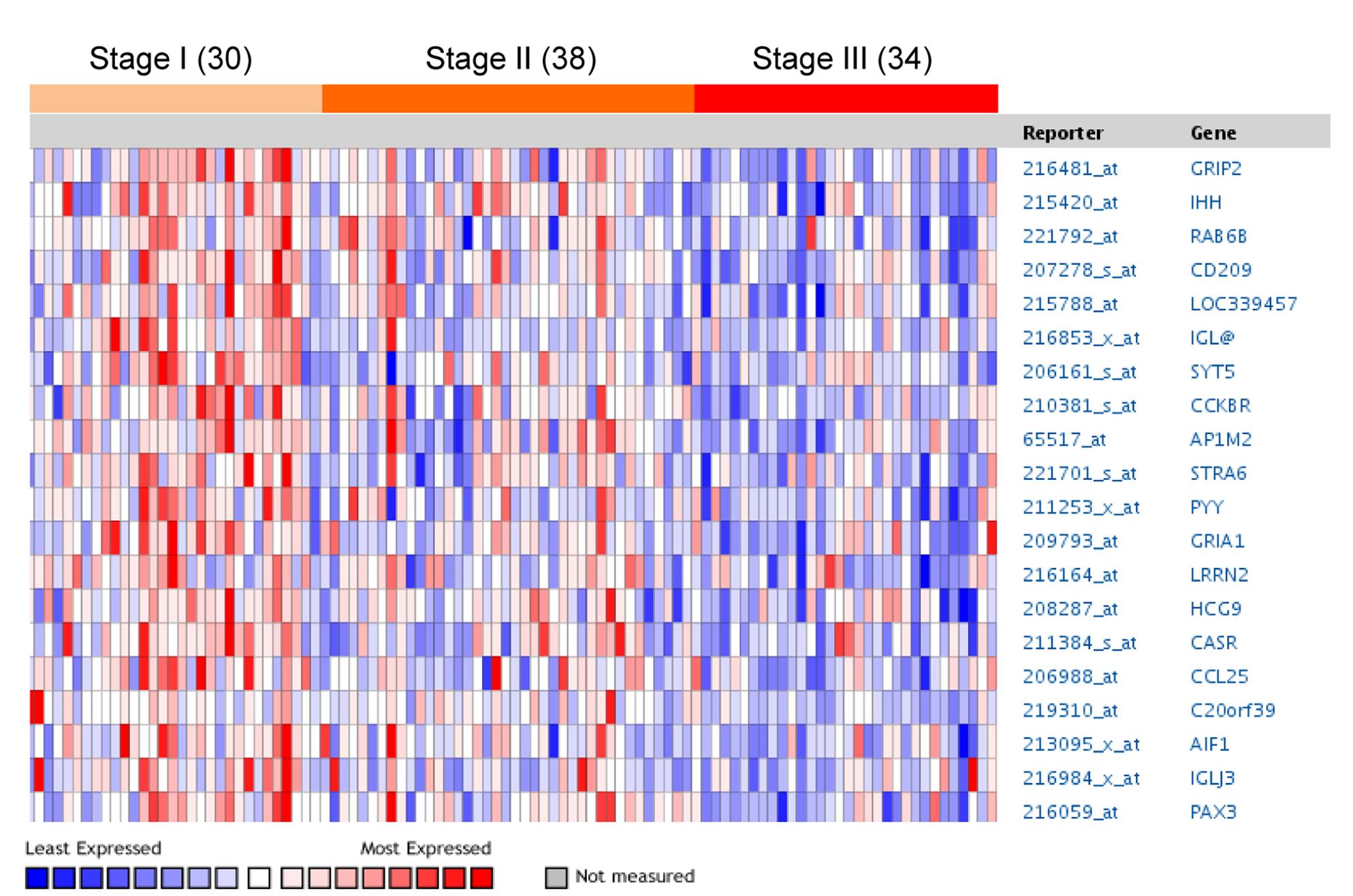
Tableau représentant le niveau d'expression de certains gènes de patients atteints de myélome multiple, en fonction du stade plus ou moins avancé de la maladie (stage 1 à 3). Le rouge indique une forte expression, le bleu une faible expression. Plus le stade de la maladie est avancé, plus les gènes s'éteignent. © Université d'Uppsala / Plos One
Modifications épigénétiques par PRC2
Les chercheurs ont ensuite corrélé ces résultats à des données génomiquesgénomiques déjà publiées auparavant. Ils ont pu montrer que beaucoup des gènes dérégulés sont également la cible des protéinesprotéines Polycomb (PcG). Ces protéines forment des complexes PRC1 ou PRC2 qui peuvent remodeler la chromatinechromatine (forme de l'ADNADN soumise à des modifications chimiques pour réguler son expression) afin d'éteindre certains gènes de manière épigénétique.
Il existe plusieurs modifications possibles de la chromatine, mais chacune porteporte la marque spécifique d'une enzymeenzyme particulière. Ainsi, PRC2 forme des tri-méthylationsméthylations de l'histonehistone H3, sur l'acide aminéacide aminé lysinelysine 27 (H3K27me3). L'immunoprécipitation de la chromatine, une méthode qui consiste à récupérer spécifiquement un morceau d'ADN qui contient la modification épigénétique d'intérêt, a permis de montrer que les gènes sous-exprimés dans les cellules cancéreuses possèdent davantage de tri-méthylations typiques de PRC2 que les mêmes gènes des cellules saines. PRC2 est donc bien responsable de l'extinction pathologiquepathologique des gènes analysés.
L'applicationapplication de moléculesmolécules chimiques nommées DZNep et LBH589, a permis d'une part de réactiver les gènes éteints par PRC2, mais aussi d'éliminer un composant actif du complexe PRC2, ce qui diminue la prolifération et augmente l'apoptoseapoptose des cellules cancéreuses. Ces résultats observés sur des lignées cellulaires ont ensuite été confirmés sur le modèle murinmodèle murin du myélome multiple : le traitement de ces souris malades par la molécule LHB589, a permis de prolonger significativement la vie des rongeursrongeurs, par rapport à des souris non traitées.
Maintenant que le mécanisme moléculaire de la mise en place de ce cancer est mieux compris, de nouvelles molécules pourront être développées pour cibler spécifiquement PRC2, afin de réduire son activité et de redonner aux gènes touchés une expression normale. Les patients atteints de myélome multiple pourront peut-être bientôt, grâce à ces travaux, vivre plus longtemps.

par Claire Peltier, Futura
le 21 juillet 2010























