






au sommaire


Analyse chez Arabidopsis thaliana
3.1. Analyse avec RSA-tools :
3.1.1. Utilisation d'oligo-analysis :
En utilisant comme paramètre la fréquence des différents oligonucléotidesoligonucléotides dans les régions non codantes du génomegénome d'A. thaliana, le programme oligo-analysis analyse l'échantillon de promo-teur en comparant les différents hexanucléotides présents par rapport à leur fréquence dans le gé-nome non codant complet d'A. thaliana. L'application aux promoteurs des gènesgènes RSA d'A. thaliana donne les résultats du tableau 2.
Tableau 2.Résultat d'oligo-analysis
Une séquence consensus est par définition l'alignement optimal de séquences similaires plus ou moins répétées dans une portion de génome. Or les séquences révélées dans le tableau 2 sont rela-tivement similaires. De ce fait oligo-analysis propose de les assembler afin de déterminer un consensus (tableau 3) ATGCTGCAA et son complémentaire TTGCAGCAT.

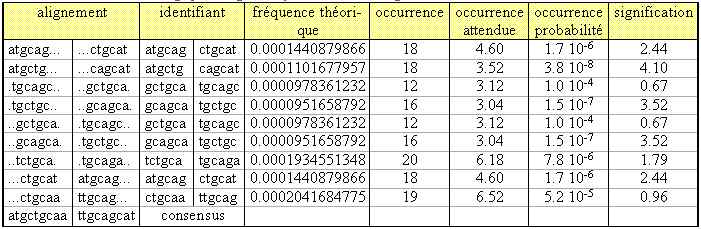
Tableau 3. Assemblage par oligo-analysis du cluster ctgcaa
La visualisation sur la figure 2 de ces hexanucléotides dans les séquences promotrices considérées, montre que plusieurs d'entre eux correspondent à un même site et résultent de leur similarité à une base prés. La modification de la visualisation, aboutit à la figure 3 où seuls les sites sont visibles. La visualisation de ces sites permet de voir l'existence de plusieurs boîtes par promoteur, concentrées essentiellement au delà de -1000, 0 étant le début de la séquence codante.

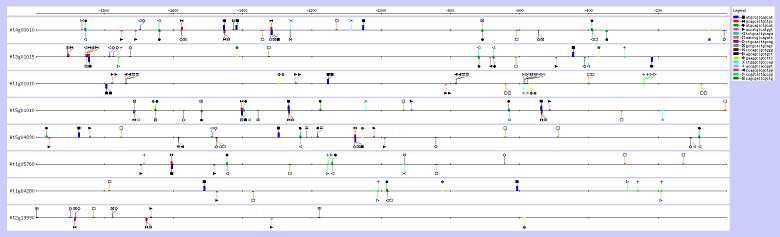
Figure 3. Visualisation de l'ensemble des hexanucléotides

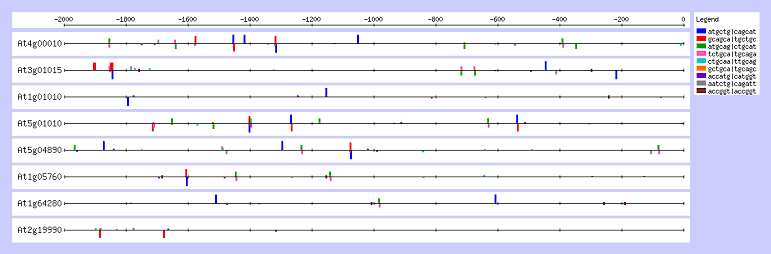
Figure 4. Visualisation de l'ensemble des sites
3.1.2. Utilisation de consensus :
L'analyse matricielle après 1000 cycles aboutit à l'élaboration de 4 matrices dont le score est signi-ficatif, répertoriées dans le tableau 4. Ces matrices donnent 4 consensus définis mathématiquement qui sont tous différents de celui déterminé par l'analyse précédente. Elles définissent un ensemble de sites visibles sur la figure 4. La comparaison avec la figure 3, montre que ces sites sont proches avec ceux trouvés par oligo-analysis, différents de quelques bases dans leurs positions ainsi que dans leurs séquences.

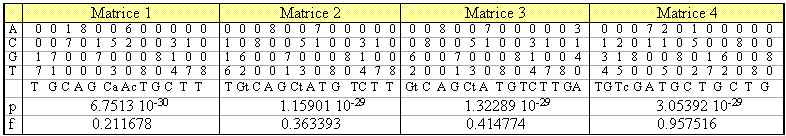
Tableau 4. Résultat de consensus

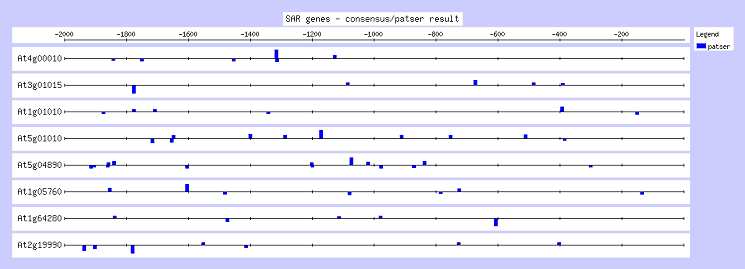
Figure 5. Visualisation de l'analyse par consensus
3.1.3. Application d'une fenêtre flottante :
Le principe de la fenêtrefenêtre flottante est de déplacer le long de la séquence à analyser une règle de lonlon-gueur fixe afin de déterminer la concentration des séquences qui permettent de déterminer celle consensus dans cette longueur. En prenant une longueur de 50 bases, le programme sliding window donne la figure 6, montrant ainsi la présence de boîtes, plus ou moins importantes, dont la localisa-tion est essentiellement entre -1000 et -2000 et de plus recouvre les positions déterminées par l'analyse avec oligo-analysis.
La comparaison entre les figures 4 et 6 permet de supposer que 2 à 3 boîtes contenant au moins 2 sites, formant le consensus, permettent à un facteur de transcriptiontranscription d'activer le promoteur.


Figure 6. Visualisation de l'analyse en fenêtre flottante de 50pb
3.2. Analyse avec Motif Sampler :
L'analyse et la comparaison des séquences promotrices des gènes SAR peuvent aussi être effec-tuées par chaîne de Markov de façon similaire par l'intermédiaire du site Motif Sampler (Thijs et al., 2001).

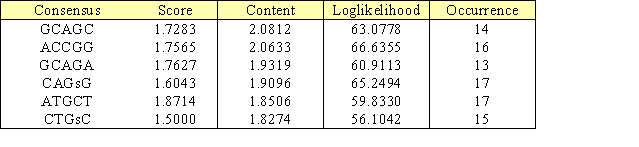
Tableau 5. Résultat général de motif sampler
La différence d'algorithme avec RSAalgorithme avec RSA tools donne néanmoins un consensus proche de celui trouvé par oligo-analysis : GCAGC.
3.3. Interprétation de l'ensemble des résultats :
La suite de programme RSA tools aboutit à la définition d'une séquence consensus sur l'ensemble des promoteurs considérés. Classé par ordre d'occurrences significatives pour Oligo-analysis, la plus élevée, 4,1, se retrouve dans toutes les séquences et correspond à TGCAGC GCAGC TTGAC TGACG. De plus Motif Sampler donne un résultat similaire, GCAGC.
Or cet hexanucléotide est relativement similaire à TTGAC définissant la famille de facteurs de transcription WRKY (Maleck et al., 2000). Mais ce consensus se trouve être très proche de TGACG qui définit lui la famille TGA bZIP (Després et al., 2000).
Parmi cette famille deux protéinesprotéines du système de résistancerésistance acquise forment le dimère NPR1/NIM1. Ce dernier a la propriété d'activer la transcription du gène PR1 via la séquence TGCAG (Després et al., 2000). Par conséquent le consensus déterminé pour ces promoteurs d'Arabidopsis thalianaArabidopsis thaliana pourrait correspondre à celui reconnu par le facteur de transcription NPR1/NIM1.
L'ablationablation partielle du promoteur du gène PR1 montra que la boîte principale d'activation se trou-vait entre -645 et -640 or il se trouve que la séquence en question, ACGTCA, est reconnue par les TGA-bZIP et dont le complément est TGACGT (Lebel et al., 1998). L'analyse effectuée par RSA-tools ne détecte pas cette boîte mais d'autres dont les plus proches sont entre -593 et -582 ainsi qu'entre -1279 et -1268. Les études moléculaires sur le promoteur de PR1 confirment donc la fiabi-litélité de ce consensus. Néanmoins d'autres seront nécessaires afin de déterminer l'importance du nombre de boîte.
La présence sur le promoteur de PR1 de ces deux types de boîtes d'activation, suggère que ce gène intervient dans deux systèmes différents nécessitant des protéines communes à la réponse systémi-que acquise (TGA-bZIP) et à la réponse au stress oxydatifstress oxydatif (WRKY). Dans le cas de l'activation par NPR1/NIM1, il est probable que celle par le facteur du type WRKY soit inhibé directement ou via une autre protéine, SNI-1 (Rowland et al., 2001).
De ces deux méthodes ressort donc un consensus du type TGA bZIP reconnaissable par NPR1/NIM1 constitutif. Or le gène NPR1 (NIM1 étant la dénomination résultant de la surexpres-sion) fait parti de l'échantillon considéré, il se pourrait donc que l'activation du système de résis-tance acquise résulte d'une auto-amplification de ce dimère afin de dépasser un seuil critique d'activation.

par Emmanuel Poulouin
le 25 février 2003



. © SPF Affaires étrangères, commerce extérieur et coopération au développement")









