

Le diabète correspond à un excès durable de la concentration de glucose dans le sang et peut entraîner des complications de santé à long terme. Pour mieux comprendre la pathologie, une équipe de chercheurs français étudie depuis plusieurs années le rôle de la leptine, une hormone impliquée dans le contrôle de l’appétit qui transmet au cerveau le signal de satiété. Outre ces recherches approfondies, l'équipe a réussi une autre prouesse.
au sommaire
Dans 90 % des cas, il s'agit de diabète de type 2 (DT2). Les patients qui en sont atteints sont généralement obèses ou en surpoids, et les facteurs de risque de déclenchement de la maladie incluent la sédentarité ainsi qu'une alimentation déséquilibrée. C'est une équipe de chercheurs de l'Inserm, d'Université de Lille et du CHU de Lille au sein du laboratoire Lille Neuroscience et Cognition 1 qui étudie depuis plusieurs années le rôle de la leptine, une hormone impliquée dans le contrôle de l'appétit, qui transmet au cerveau le signal de satiété. Dans une nouvelle étude publiée dans la revue Nature Metabolism, en plus d'approfondir les connaissances scientifiques sur le mécanisme de satiété, les chercheurs sont parvenus à reproduire chez la souris un nouveau modèle de diabètediabète utile et pertinent pour faire avancer la recherche sur la maladie.
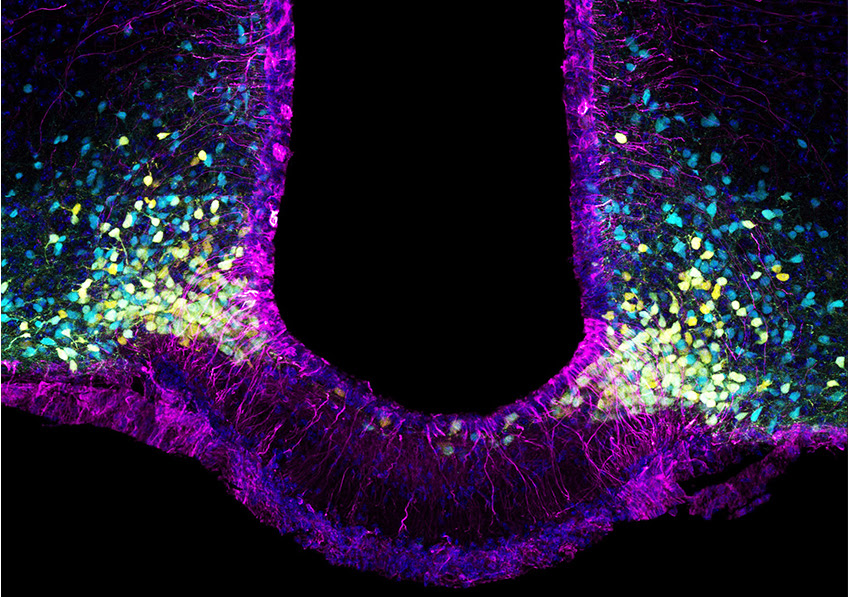
La leptine (« hormone de la satiétéhormone de la satiété » ou « hormone coupe faim ») est une hormone sécrétée par le tissu adipeuxtissu adipeux, proportionnellement aux réserves de graisses dans l'organisme, qui régule l'appétit en contrôlant la sensation de satiété. Elle est transportée vers le cerveau par des cellules appelées tanycytes, dans lesquelles elle entre en s'arrimant à des récepteurs appelés LepR.
Les tanycytes sont donc la porteporte d'entrée de la leptine vers le cerveau, aidant cette hormone à franchir la barrière hémato-encéphaliquebarrière hémato-encéphalique et à délivrer aux neuronesneurones l'information de satiété. De précédentes recherches ont révélé que le transport de la leptine est altéré chez les sujets obèses et en surpoids, expliquant en partie des dysfonctionnements dans la régulation de l'appétit, puisque l'information de satiété parvient plus difficilement à atteindre le cerveau. Dans leur nouvelle étude, les chercheurs se sont intéressés de plus près à ce transport de la leptine jusqu'au cerveau, et plus précisément au rôle des récepteurs LepR.

Quel est le rôle des récepteurs de l’hormone de la satiété dans la gestion du glucose ?
Dans des modèles de souris, les chercheurs ont retiré le récepteur LepR situé à la surface des tanycytes. Au bout de trois mois, les souris ont enregistré une forte augmentation de leur massemasse grasse (multipliée par deux sur la période) ainsi qu'une perte de masse musculaire (diminuée de plus de la moitié). La prise de poids totale n'a été qu'assez modérée. Les scientifiques ont par ailleurs mesuré régulièrement la glycémie des animaux après injection de glucoseglucose. Ils ont constaté que, pour maintenir une glycémie normale (entre 0.70 et 1.10 g/L), les souris ont sécrété davantage d'insulineinsuline au cours des quatre premières semaines de l'expérience.
Trois mois après le retrait du récepteur, leur capacité de sécrétionsécrétion d'insuline par le pancréas semblait épuisée. Le fait de retirer les récepteurs LepR et d'altérer le transport de la leptine vers le cerveau a donc conduit les souris à développer dans un premier temps un état pré-diabétiquediabétique. Celui-ci survient lorsque l'organisme libère de l'insuline en plus grande quantité que d'accoutumée pour contrôler la glycémieglycémie. Puis, à plus long terme, les souris deviennent incapables de sécréter de l'insuline et donc de contrôler la quantité de glucose présente dans le sang. Ces données suggèrent ainsi qu'un transport altéré de la leptine vers le cerveau, via les récepteurs LepR, est impliqué dans le développement du diabète de type 2.
Le saviez-vous ?
Chez un animal ou un individu normal, la glycémie augmente légèrement après ingestion de glucose et redescend rapidement. En effet, pour retrouver des valeurs normales, le pancréas sécrète de l’insuline qui aide le glucose à pénétrer les cellules de l’organisme. Chez l’animal privé du récepteur LepR à la porte d’entrée de la leptine dans le cerveau, la glycémie est anormalement élevée à jeun et à fortiori après ingestion du glucose. Le pancréas devient incapable de sécréter de l’insuline nécessaire à la pénétration du glucose dans l’organisme. La « surdité » du cerveau à l’information véhiculée par la leptine rend ainsi le pancréas non fonctionnel.
Ce travail a été conduit en collaboration avec deux laboratoires de l'Institut Cochin et de l'Université de Strasbourg dans le cadre d'un projet financé par l'Agence Nationale de la Recherche et deux laboratoires Européens, l'un à l'Université de Lübeck en Allemagne et l'autre à l'Université de Saint Jacques de Compostelle en Espagne, dans le cadre d'un financement de la Communauté Européenne. Par ailleurs, le laboratoire Lille Neuroscience et Cognition est membre du LabEx Egid (European Genomic Institute for Diabetes), mais aussi de Distalz (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimerALZheimer's disease).
Dans la dernière partie de leurs travaux, les chercheurs ont procédé à la réintroduction de la leptine dans le cerveau et ont constaté une reprise immédiate de son action favorisant la fonction du pancréaspancréas et notamment sa capacité à sécréter de l'insuline pour réguler la glycémie. Les souris ont retrouvé rapidement un métabolismemétabolisme en bonne santé.
Cette étude met donc en lumièrelumière le rôle du cerveau dans le diabète de type 2 et contribue par ailleurs à faire évoluer la recherche sur la maladie, qui n'était pas considérée jusqu'alors comme une maladie du système nerveux centralsystème nerveux central. « Nous montrons en effet d'une part que la perception de la leptine par le cerveau est indispensable pour la gestion de l'homéostasiehoméostasie énergétique2 et de la glycémie. D'autre part, que le blocage du transport de la leptine vers le cerveau altère le bon fonctionnement des neurones qui contrôlent les sécrétions d'insuline du pancréas », conclut Vincent Prévot, directeur de recherche à l'Inserm, dernier auteur de l'étude.
Vers une meilleure compréhension d'un autre diabète moins étudié
Autre résultat intéressant de cette étude : en retirant le récepteur LepR à la porte d'entrée de la leptine dans le cerveau, le modèle animal obtenu présente les caractéristiques de ce qu'on appelle le « Diabète Est Asiatique » encore peu étudié par les chercheurs. Ce phénotypephénotype de diabète concerne principalement les populations de Corée et du Japon.
Alors que le « Diabète Occidental » est la plupart du temps associé à un surpoids marqué (IMCIMC >25) ou à une obésitéobésité morbide (IMC >30) », cet autre phénotype de diabète de type 2 présente souvent un léger surpoids, une augmentation de la graisse abdominale et une insuffisance en insuline liée à une défaillance de la sécrétion d'insuline par le pancréas. Selon les chercheurs, le développement de ce nouveau modèle animal permettra de faire avancer la recherche sur cette pathologiepathologie qui touche des millions de personnes.
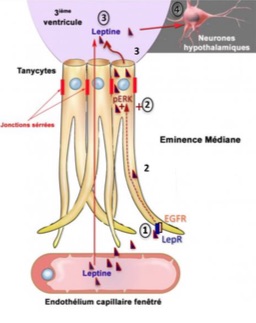
. Ces cellules capturent la leptine circulante à partir des vaisseaux sanguins qui, à cet endroit-là, ont la particularité de la laisser passer (étape 1). Lors de son parcours dans le tanycyte, la leptine capturée par le LepR active le récepteur à l’ « EGF » (ou EGFR) qui lui-même active une voie de signalisation « ERK » (étape 2) qui enclenche sa libération dans le liquide céphalorachidien (étape 3). La leptine active alors les zones cérébrales qui véhiculent son action anorexigène (« coupe-faim »), mais aussi le contrôle du fonctionnement du pancréas (étape 4). © Vincent Prévot")

par INSERM
le 8 août 2021






















